Coflex
The coflex(TM) device
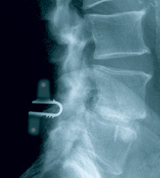
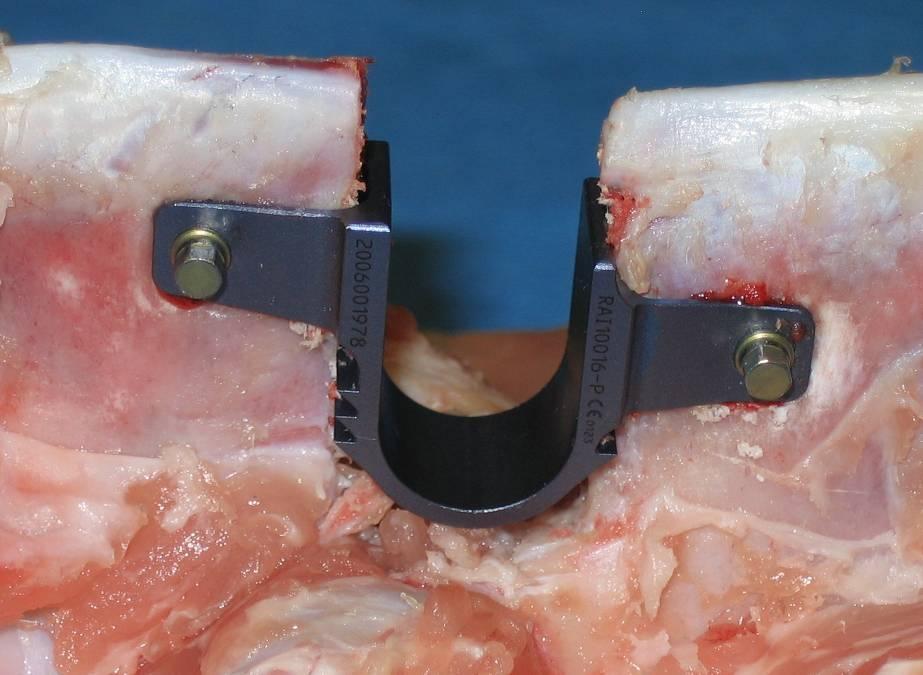
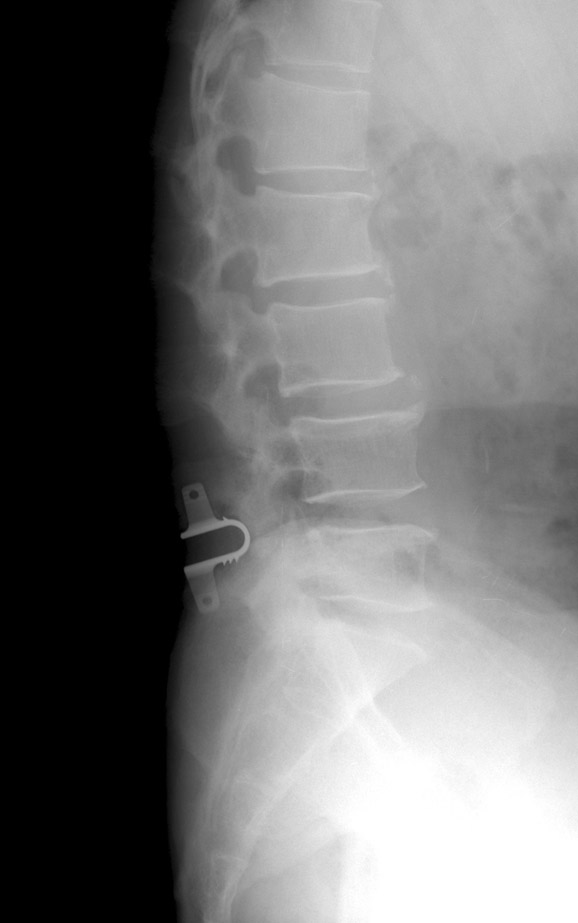
The coflex(TM) device is a U-shaped titanium alloy, available in 5 sizes from 8mm to 16mm. It is indicated for use in patients with radiographically confirmed moderate to severe stenosis with neural element compromise resulting in claudication and/or radicular symptoms isolated to 1 or 2 levels, in the region of L1 to L5.
The coflex™ interspinous implant offers a simple surgical treatment strategy with a low risk potential. Not a single patient was dissatisfied with the outcomes of their surgery,” Dr. Bertagnoli reported.
The coflex interspinous implant was invented by Dr. Jacques Samani in 1994 and has been in continuous use since 1995 outside the United States. Initially, the product was known as the Interspinous “U” and was marketed by Fixano SAS (Peronnas, France). Transfer of ownership to Paradigm Spine was finalized in early 2005, and the product was renamed “coflex interspinous implant”. The design and materials were not changed, but two new sizes were added. The only differences between the Interspinous “U” and the coflex interspinous implant are the manufacturing technique (from a wire EDM manufactured device to a milled device) and the tightening of tolerances. Surgeons with a significant history and patient volume were contacted to participate in a retrospective review of their patient experience with the Fixano Interspinous “U”, the predecessor device to the coflex interspinous implant. Four sites participated in this data collection effort. The surgeons attempted to contact all patients who had received the device. All patients who were greater than six months postoperative were given the option to participate in this data collection, which the company believes helped to minimize selection bias. Of the 589 patients identified by the surgeons, 429 (73%) responded and agreed to participate. All patients were asked to return for contemporaneous history and clinical examination and dynamic x-rays. These results were compared to available patient records pertaining to their quality of life and neurological function and pre-existing x-rays to ascertain implant survivorship. Patient data for the retrospective study was gathered via a questionnaire that captured the following information:
1) Date of Birth
2) Gender
3) Preoperative diagnosis
4) Preoperative clinical evaluation
5) Previous conservative therapy
6) Previous spinal therapies
7) Concomitant medical conditions
8) Operative data
9) Radiographic and diagnostic tests
10) Postoperative clinical examination
11) Qualitative postoperative x-ray analysis. Of these patients, 209 were treated for spinal stenosis at a single level or two adjacent levels. This population is substantially similar to the population for the protocol that is the subject of a current USA FDA IDE.